Carbetocin is a functional selective Gq agonist that does not promote oxytocin receptor recycling after inducing β-arrestin-independent internalisation
Ilaria Passoni1, Marianna Leonzino2, Valentina Gigliucci2, Bice Chini2,3 and Marta Busnelli1,2
1 Department of Medical Biotechnology and Translational Medicine, Università degli Studi di Milano, Milan, Italy 20129
2 CNR, Institute of Neuroscience, Milan, Italy 20129
3 Humanitas Clinical and Research Institute, Rozzano, Italy, 20089
ABSTRACT
Carbetocin, a long-acting oxytocin analogue, has been reported to elicit interesting and peculiar behavioural effects. The aim/purpose of this work was to investigate the molecular pharmacology of carbetocin in order to better understand the molecular basis of its action in the brain. Using Bioluminescence Resonance Energy Transfer (BRET) biosensors, we characterised the effects of carbetocin on the three human oxytocin/vasopressin receptors expressed in the nervous system: the oxytocin receptor (OXTR) and the vasopressin V1a (V1aR) and V1b (V1bR) receptors. Our results indicate that 1) carbetocin activates the OXTR but not the V1aR and V1bR at which it may act as an antagonist; 2) carbetocin selectively activates only the OXTR/Gq pathway displaying a strong functional selectivity; 3) carbetocin is a partial agonist at the OXTR/Gq coupling; 4) carbetocin promotes OXTR internalisation via a previously unreported β-arrestin-independent pathway; 5) carbetocin does not induce OXTR recycling to the plasma membrane. All together, these molecular pharmacology features identify carbetocin as a substantially different analogue as compared to the endogenous oxytocin and, as a consequence, carbetocin is not expected to mimic oxytocin in the brain. Whether these unique features of carbetocin could be exploited therapeutically remains to be established.
Keywords: carbetocin, oxytocin receptor, vasopressin receptor, β-arrestins, receptor recycling
INTRODUCTION
In the CNS, oxytocin acts as a neurotransmitter/neuromodulator regulating several aspects of social behaviour, learning and memory processes, stress and anxiety responses (6-7). On the basis of its capability to promote social interactions in all vertebrates, including humans, oxytocin has been proposed as a clinical treatment to relieve social impairments associated with neurodevelopmental and psychiatric disorders (8-9). In the past decade, a number of clinical trials have been performed in autism and schizophrenia but, unfortunately, no consensus on the real efficacy of oxytocin on these conditions has yet been reached (10). Several factors contribute to the limited/controversial efficacy of oxytocin in the clinical practice: 1) oxytocin has a short half-life in the plasma (11) and in the cerebrospinal fluid (12) and, as a consequence, its pharmacological effects are short lived; 2) oxytocin does not cross the blood-brain barrier in a significant amount (12) and thus, when given by parenteral administration, it does not reach the brain; 3) intranasal oxytocin administration, a way to deliver exogenous oxytocin directly into the brain, has not yet been proven to do so (13), leading to very poorly defined pharmacokinetics parameters (doses, intervals, metabolites); 4) oxytocin binds to and activates the vasopressin V1a and V1b receptor subtypes, that are also highly expressed in the brain where they promote different and even opposite effects from the oxytocin receptor (OXTR) (14); 5) oxytocin promotes OXTR coupling to a number of different G-protein subtypes and β-arrestins (15-16), leading to the activation of multiple signalling pathways, whose precise roles within the brain are currently unknown.
To fully exploit the pharmacological potential of the OXTR and, at the same time, to overcome the limits of endogenous oxytocin, a number of oxytocin analogues have been developed through the years (17). In particular, in an attempt to increase the half-life of the hormone, the deamino-1-monocarba-(2-O-methyltyrosine)-oxytocin (Carbetocin) was synthesised by deaminating the N-terminus and by replacing the disulfide (S-S) bridge between Cys 1-6 with a CH2-S bond which connects a butyric acid functional group, at the N-terminus, and Cys 5. These modifications resulted very effective in protecting carbetocin from aminopeptidase degradation and disulfidase cleavage (18), thus increasing the half-life of this oxytocin analogue in the peripheral circulation (carbetocin 85-100 min vs. oxytocin 3.4 min) (11, 19). This increase in half-life was accompanied by an increased efficacy in vivo, as demonstrated by animal studies in which carbetocin resulted able to induce prolonged uterine contraction (20) and milk let-down activity (21). Similarly, it has been reported that, in humans, carbetocin had a prolonged effect on post- partum uterine activity in terms of both a higher amplitude and frequency of contractions in comparison to oxytocin (22). Concerning carbetocin’s central effects, studies in rodents demonstrated that carbetocin reduced anxiety-like behaviours in the elevated plus maze test (23), had antidepressant-like effects in the forced swimming test (24) and attenuated the negative emotional consequences of opioid withdrawal (25-26).
Interestingly, behavioural studies that directly compared carbetocin and oxytocin, have reported different and, in some cases opposite, effects. Differently from oxytocin, carbetocin failed to revert the deficits in Pre-Pulse Inhibition of the startle reflex (PPI) in Brown Norway rats, a rat strain that has naturally low PPI, thus suggesting that carbetocin does not have antipsychotic-like central effects (27). In the open field paradigm, while oxytocin induced a reduction in exploratory activity and increased grooming time, carbetocin had a slightly increasing effect on exploratory activity and no effect on grooming (28). Moreover, in rats previously exposed to restraint stress, oxytocin reduced locomotion and increased grooming, while carbetocin increased locomotion and decreased grooming (29). Finally, carbetocin but not oxytocin had long-term ameliorating effects on restraint stress-induced behavioural changes (30).
The aim of this study was to investigate in details the pharmacological properties of carbetocin on the OXTR and vasopressin V1a and V1b receptors, in order to identify the molecular mechanisms which could account for the differences observed between carbetocin and oxytocin at the behavioural level. We found that carbetocin is highly selective for the OXTR as compared to the vasopressin V1a and V1b receptors, and that it specifically activates only the OXTR/Gq pathway, displaying functional selective properties. Finally, we investigated the carbetocin effects on OXTR internalisation and intracellular trafficking and found that carbetocin promotes OXTR internalisation through a previously undescribed β-arrestin-independent internalisation pathway, that also negatively influences OXTR recycling.
MATERIALS AND METHODS
Peptide and Reagents
Oxytocin, carbetocin and arginine-vasopressin were obtained from Bachem. Coelenterazine h came from Molecular Probes, Invitrogen, and coelenterazine 400a (CLz400) from Biotium.
Cell Culture
HEK293 cells were maintained in DMEM supplemented with 10% FBS, 200 U/ml penicillin, 200 mg/ml streptomycin, and 2 mM L-glutamine (all purchased from Sigma-Aldrich).
cDNA Constructs
The expression vectors for G-proteins fused to Renilla luciferase Gαq-97-Rluc8, Gαi1-91-Rluc8, Gαi2- 91-Rluc8, Gαi3-91-Rluc8, Gαo-91-Rluc8 and the vectors for GFP10-Gγ2 and Gβ1 were a gift from Dr. Celine Gales (Inserm U858, Toulouse, France) and are described in (31). The plasmid encoding for the human OXTR (hOXTR) and the hOXTR-Rluc vectors were the same as in (15). The plasmid for V1aR and V1bR were a gift from Prof. Guillon and Dr. Mouillac (IGF, CNRS, Montpellier, France). The expression vectors for β-arrestins-Venus were kindly provided by Prof. Locati (Humanitas Research Hospital Rozzano, Italy). The hOXTR-RFP construct was generated amplifying the entire coding sequence of hOXTR by PCR and using the forward primer 5’- CAAACTCGAGA TGGAGGGCGCGCTCGCAG-3’ and the reverse primer 5’- GTTTGGATCCGCCGTGGATGGCTGGGAG-3’ and the Pfu DNA polymerase (Promega). The resulting PCR product was subcloned into the Tag-RFP plasmid (Evrogen) using the XhoI and BamHI restriction sites. hOXTR-RFP fusion constructs were sequenced on both DNA strands (MWG Eurofins).
Transfections
For Bioluminescence Resonance Energy Transfer (BRET) experiments, cells were seeded at a density of 3,1 x 106 cells/well in 100-mm plates and transfected after 24 h with polyethyleneimine (PEI linear, Mr 25,000, Polysciences) as previously described in (15). 24 h after transfection, the supplemented DMEM was renewed, and the cells were cultured for a further 24 h before the experiments. 48 h after transfection, the cells were washed twice, harvested, and resuspended with PBS supplemented with 0.1% (w/v) glucose at room temperature.
For imaging experiments, cells were seeded on glass coverslips (3 x 105 cells/coverslip), allowed to grow for 24 h (60% confluence), and transfected with TurboFect (Thermo Scientific). For each transfection sample, 1 μg of cDNA was mixed with 3 μl of TurboFect in 400 μl of DMEM supplemented with 2 mM L-glutamine and incubated for 15 min at room temperature. DNA/TurboFect complexes were gently added to the cells in 3,6 ml of complete medium supplemented with 10% FBS. 24 h after transfection, the supplemented DMEM was renewed, and the cells were cultured for a further 24 h before the experiments.
BRET Assays
To screen for the effects of the different ligands on G-protein activation, we performed BRET2 assays. Plasmids encoding for GFP10-Gγ2, Gβ1, and the hOXTR or vasopressin receptors (V1aR and V1bR) were co-transfected together with the different Gα-Rluc8 plasmids in HEK293 cells. To study OXTR/β-arrestin interactions we used kinetic BRET1 experiments and the cells were co- transfected with hOXTR-Rluc and β-arrestin1-Venus or β-arrestin2-Venus. 48 h after transfection, cells were washed twice, harvested, and resuspended in PBS-glucose 0.1% (w/v) at room temperature. The protein content of cells was determined using DC Assay (Biorad) and cells were resuspended to the final protein concentration of 1 mg/ml. Cells (80 μg of proteins/well) were then distributed in a white 96-well microplate (Optiplate, PerkinElmer Life Sciences). For the G-protein activation studies, cells were incubated for 2 min with the indicated ligands or PBS (untreated cells) before substrate addition. BRET2 energy transfer between Rluc8 and GFP10 was measured immediately after the addition of the Rluc8 substrate coelenterazine 400a (5 μM, Biotium), using an Infinite F500 reader plate (Tecan) that allows the sequential integration of light signals detected with two filter settings (Rluc8 filter, 370-450 nm; GFP10 filter, 510-540 nm). The data were recorded, and the BRET2 signal was calculated as the ratio between GFP10 emission and the light emitted by Rluc8. The changes in BRET signal induced by the ligands were expressed on graphs as “BRET ligand effect” using the formula
BRET ligand effect = (emission GFP10ligand/emission Rluc8ligand) - (emission GFP10PBS/emission Rluc8PBS)
To study the kinetics of the OXTR-β-arrestin interactions, coelenterazine h (the substrate specific for BRET1 experiments, Molecular Probes, Invitrogen) was added to cells 8 min before the addition of oxytocin (10 μM) or carbetocin (10 μM), and readings were registered using an Infinite F500 reader plate (Tecan) and filter set (Rluc filter, 370-480 nm; Venus filter, 520-570 nm). To determine the half-time (t1⁄2) of oxytocin- and carbetocin-induced BRET, the data were recorded as the difference between the “ligand-promoted BRET” signal and the average of the baseline (PBS- treated) BRET signal, and the time at which the half-BRET peak was reached was estimated.
Fluorescence Microscopy, β-arrestin Recruitment, Internalisation and Recycling Studies
In β-arrestin recruitment studies, cells were co-transfected with hOXTR-RFP and β-arrestin1- or β- arrestin2-Venus constructs and after 48 h were preincubated for 30 min in serum-free medium at 37°C and treated with oxytocin (100 nM) or carbetocin (1 μM) for 2 min. In the internalisation and recycling studies, cells were transfected with hOXTR-RFP. 48 h after transfections, cells were preincubated for 30 min in serum-free medium at 37°C and treated with oxytocin (100 nM) or carbetocin (1 μM) for 15 min. In the recycling experiments, agonist was removed after a period of 15 min with an acid wash pH 3.3 (150 mM NaCl, 5 mM CH3COOH) and the cells were incubated for a subsequent period of time of 4 h in serum-free medium. All the cellular processes were blocked by placing the dishes on ice and cells were immediately processed for fluorescence microscopy analysis. Cells seeded on glass coverslips were washed twice with 10 mM sodium phosphate buffer, pH 7.4, containing 150 mM NaCl [high-salt buffer (HS)], and fixed for 20 min at room temperature with 4% (w/v) paraformaldehyde. Fixed cells were stained with DAPI, washed five times with HS, then once with H2O. Glass coverslips were mounted on glass slides with MOWIOL and analysed with an Axiovert 200M (Zeiss) confocal system equipped with a spinning disc (Perkin Elmer Life Sciences) with a 63X objective.
Statistical Analysis
All data were analysed using Graph-Pad Prism 5.0 software. BRET data are all expressed as mean ± SEM, calculated by simultaneous analysis of at least three different experiments performed at least in duplicate. Ligand-induced BRET ratios were analysed with one-way ANOVA followed by Dunnett’s post-hoc test, comparing oxytocin, carbetocin or arginine-vasopressin with vehicle within each G-protein subtype, to determine statistically significant differences in treatments (*p<0.05, **p<0.01, ***p<0.001). Concentration-response BRET curves were analysed by means of nonlinear curve fitting using the sigmoidal dose-response equation. BRET kinetics data were normalised by setting the zero time point immediately after the addition of the ligand, and the data were analysed by means of nonlinear least-squares fitting to the one-phase exponential association equation.
RESULTS
Carbetocin differs from oxytocin, being a “functional selective” OXTR/Gq agonist
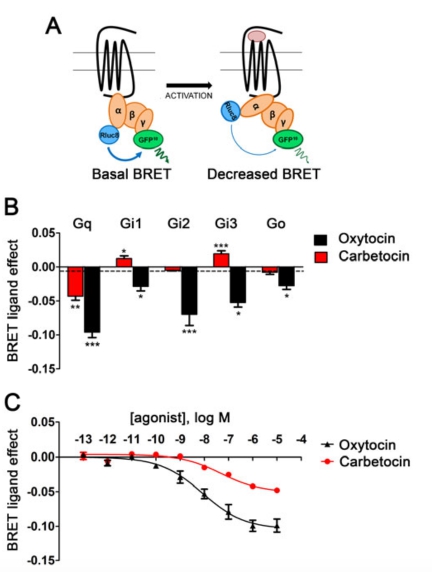
We have previously demonstrated that the native peptide oxytocin can activate OXTR coupling to different G-protein subtypes: Gq, Gi and Go (15).
To investigate the coupling specificity of human OXTR (hOXTR) in response to carbetocin, we employed BRET-based biosensors (Fig. 1A). HEK293 cells were transfected with the hOXTR in combination with different Gα subunits fused to the BRET energy donor Renilla reniformis luciferase (Gα-Rluc8), and with the energy acceptor, a blue-shifted variant of Aequorea Victoria green fluorescent protein (GFP10), N-terminally fused to the Gγ2 subunit (GFP10-Gγ2) (30). Ligand-induced OXTR activation of the G-protein leads to GDP release coupled to a conformational rearrangement of the heterotrimeric complex, and, as a consequence, to a decrease in the BRET ratio (Fig. 1A). To screen oxytocin and carbetocin for their efficacy to activate OXTR signalling, we used a 10 μM dose, that for oxytocin is known to maximally activate all the different G-protein subtypes (15) and that for carbetocin is 1,000 fold greater than its affinity for the OXTR. For all the G-proteins analysed, one-way ANOVA showed an effect of treatment on OXTR: Gq [F(2,51)=52.57, p<0.0001]; Gi1 [F(2,17)=10.53, p<0.01]; Gi2 [F(2,18)=204.3, p<0.0001]; Gi3 [F(2,17)=34.55, p<0.0001]; Go [F(2,40)=17.21, p<0.0001]. As previously demonstrated (14), oxytocin (10 μM) activated Gq, Gi1, Gi2, Gi3 and Go proteins; on the contrary, carbetocin (10 μM) activated only the Gq protein, indicating that carbetocin is OXTR/Gq “functional selective” analogue (Fig. 1B). Notably, for carbetocin, we observed a modest but significant increase in the energy transfer in the presence of Gi1 and Gi3 (Fig. 1B), indicating a particular rearrangement of the receptor associated with a more close organization of the trimeric G-protein complex which could indicate inverse agonism (32).
Carbetocin is a partial agonist for the OXTR/Gq coupling
Using the same Gq BRET biosensor (Fig. 1A), we performed dose-response curves. We found an increased EC50 for carbetocin: EC50= 48.8 ± 16.09 nM for carbetocin and EC50= 9.7 ± 4.43 nM for oxytocin, respectively (Fig. 1C). This right-shifted EC50 is congruent with the reported affinity for the OXTR (Ki=7 nM), which is about 10-fold lower than that of oxytocin (Ki=0.71 nM) (31). We also found that maximal activation (BRETmax) induced by carbetocin was approximately half (45± 6%) than that of oxytocin, suggesting that carbetocin is a partial OXTR/Gq agonist (Fig. 1C). This is in agreement with earlier observations (33-34) that carbetocin on isolated uterine strips generates a reduced maximal contractile response than oxytocin, indicating a “weaker” activation of the OXTR.
Carbetocin does not activate vasopressin V1a and V1b receptors and acts as a competitive antagonist
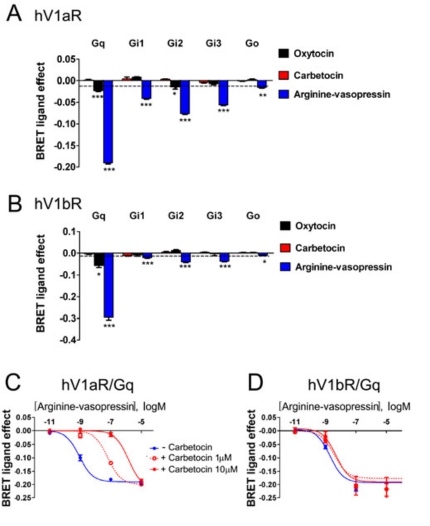
A previous study has shown that carbetocin is able to bind to vasopressin receptors in rat myometrial homogenates (34), therefore we tested the capability of carbetocin to activate human V1a and V1b receptors with the same BRET biosensors used for OXTR (Fig. 1A).
For all the G-proteins analysed, one-way ANOVA showed an effect of treatment on V1aR: Gq [F(3,33)=1387, p<0.0001]; Gi1 [F(3,36)=86.23, p<0.0001]; Gi2 [F(3,33)=129.4, p<0.0001]; Gi3 [F(3,39)=195.6, p<0.0001]; Go [F(3,28)=11.02, p<0.0001]. An effect of treatment was observed also for V1bR: Gq [F(3,24)=358.5, p<0.0001]; Gi1 [F(3,24)=30.46, p<0.0001]; Gi2 [F(3,24)=72.92, p<0.0001]; Gi3 [F(3,24)=95.75, p<0.0001]; Go [F(3,24)=22.27, p<0.001].
Even when used at a high concentration (10 μM), at least 500 fold greater than its affinity for the vasopressin receptors (34), carbetocin was completely inactive on vasopressin V1aR and V1bR (Fig. 2A and Fig. 2B). Diversely, the same high concentration (10 μM) of oxytocin selectively promoted the activation of V1aR/Gq and V1aR/Gi2 and of V1bR/Gq. Moreover, the neuropeptide arginine-vasopressin significantly promoted the coupling/activation of the vasopressin V1aR and V1bR to all G-protein subtypes. Interestingly, our data for V1bR confirmed a previous study (35) that used an in vitro luciferase-based transcription reporter gene assay and reported no activation of this receptor by carbetocin up to 10 μM. However, diversely from our study, in the same paper a partial agonism for V1aR, arbitrarily assigned to <70%, was reported (34). Unfortunately, we cannot make direct comparisons between our BRET studies and the ones in the work just cited, as the authors didn’t describe the nature of the responsive elements present in the promoter of their construct, and we cannot therefore establish with certainty which signalling pathway was activated by the peptide.
We then tested the possibility that carbetocin acts as a competitive antagonist on the vasopressin V1a (Fig. 2C) and V1b receptors (Fig. 2D). To this aim we performed dose-response curves for the activation of Gq in the presence of arginine-vasopressin and carbetocin. Our results indicated that the simultaneous incubation with carbetocin resulted in a rightward shift of the concentration- response curves of arginine-vasopressin without a noticeable change of the maximal response (Fig. 2C and 2D). Moreover, the right-shift of the arginine-vasopressin curve is dose-dependent and indicates that carbetocin behaves as a competitive antagonist on V1aR and V1bR.
Carbetocin induces OXTR internalisation without recruiting β-arrestins
Exposure to oxytocin leads to desensitization of the OXTR (36). Desensitization, whose purpose is to protect cells from overstimulation after prolonged agonist exposure, can occour very rapidly, within seconds or minutes, and is observed in the majority of GPCRs. It is a multistep phenomenon, in which the ability to respond to stimuli is switched off for varying lengths of time. The first step is the phosphorylation of the receptor, that inhibits G-protein activation, followed by the binding of specific proteins called β-arrestins, which prevent G-protein activation and promote receptor internalisation (37).
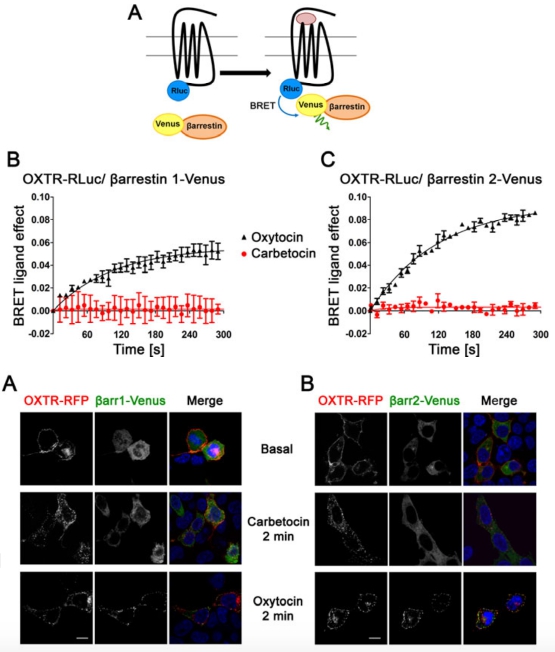
To determine whether carbetocin induced the recruitment of β-arrestins, we used a “real-time” kinetic BRET assay in which the hOXTR-RLuc construct acted as the energy donor, and the yellow variant of GFP (Venus) fused to the C-terminus of β-arrestin1 and β-arrestin2 (β-arrestin1-Venus and β-arrestin2-Venus) acted as the acceptor (Fig. 3A). In cells co-expressing hOXTR-Rluc and β- arrestin1-Venus, oxytocin at a final concentration of 10 μM, a concentration that we previously determined to be able to promote maximal β-arrestin1 and β-arrestin2 recruitment (15), increased the BRET ratio with a t1⁄2 of 119 ± 18 s; this increase remained stable for at least 5 min (Fig. 3B), thus indicating a rapid and sustained agonist-induced association between the OXTR and β- arrestin1. Similar results were obtained using the β-arrestin2-Venus construct, with a t1⁄2 of 52 ± 8 s (Fig. 3C). On the contrary, no variations in the BRET signal were observed in the presence of a high dose of carbetocin (10 μM) (Fig. 3B and 3C). These data indicate that carbetocin did not promote OXTR/β-arrestins association.
To determine if the different recruitment of β-arrestins affected OXTR internalisation, we used confocal microscopy studies. We used HEK293 cells transfected with hOXTR-RFP and β-arrestin1- Venus (Fig. 4A) or β-arrestin2-Venus (Fig. 4B). In this experiment, we stimulated cells for 2 min with 100 nM oxytocin, a concentration that is 100 fold greater than its Ki and EC50 for OXTR and that we previously verified to be able to induce the complete internalisation of OXTR without affecting its trafficking (36). Because carbetocin is characterised by Ki and EC50 for OXTR that are 10 fold bigger than oxytocin, we used for the same studies 1 μM carbetocin (38). As shown in Fig. 4, before agonist exposure (baseline) the hOXTR-RFPs were localised at the plasma membrane, while β-arrestins-Venus were homogeneously distributed into the cytoplasm. Incubation with oxytocin, but not carbetocin, induced β-arrestins accumulation at the plasma membrane that positively colocalised with OXTR, confirming that oxytocin but not carbetocin promoted the recruitment of β-arrestins. Moreover, in the presence of carbetocin and oxytocin, we observed fluorescent puncta distributed at the plasma membrane and in the cytosol, that presumably represented the OXTR internalised and localised in endocytic vesicles. All together, these results indicate that, differently from oxytocin, carbetocin promotes OXTR internalisation with a pathway independent from β-arrestins.
OXTR internalised upon carbetocin stimulation does not recycle to the plasma membrane and remains intracellular
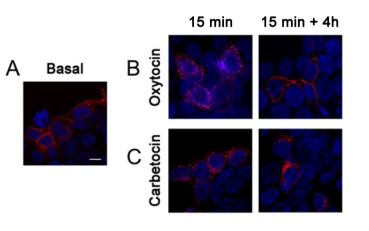
We have previously demonstrated that OXTRs endocytosed in response to 100 nM oxytocin are not sorted to the lysosomes for degradation and they are recycled back to the cell surface (36). We treated hOXTR-RFP transfected HEK293 cells (Fig. 5A, basal) with oxytocin (100 nM, Fig. 5B) or carbetocin (1 μM, Fig. 5C) for 15 min and we observed no fluorescent staining at the plasma membrane and a great number of intracellular fluorescent puncta indicating that, at this time, both agonists promoted massive OXTR internalisation (Fig. 5B and Fig. 5C). For recycling, we treated cells for 15 min, we removed the agonist with an acid wash and we maintained cells at 37°C for 4 h. In our experiments we did not block protein synthesis because we have previously demonstrated that after 4 h the OXTR de novo synthesis does not play a major role in the reappearance of the receptor at the cell surface (36). With oxytocin, in agreement with our previous observations (36), after 4 h the fluorescent staining reappeared at the plasma membrane and it was as intense as under basal conditions (Fig. 5B). On the contrary, after carbetocin OXTRs remained in intracellular vesicles and did not recycle to the plasma membrane (Fig. 5C).
These results indicate that carbetocin efficiently promotes OXTR internalisation in the absence of the recruitment of β-arrestins, and this process is not coupled to the OXTR recycling.
DISCUSSION
We report here the molecular pharmacology of carbetocin, an analogue of oxytocin originally designed to achieve a long half-life (18). In particular, we investigated 1) the coupling selectivity of carbetocin for the human OXTR; 2) the agonistic properties of carbetocin for the human OXTR, V1a and V1b receptors; 3) the carbetocin -induced OXTR internalisation and recycling. Our data indicate that carbetocin possesses specific and peculiar features in all these aspects, suggesting a pharmacological profile and therapeutic potential different from that of the endogenous neuropeptide oxytocin.
First, we investigated the coupling efficiency and selectivity of carbetocin for the human OXTR. Our data indicate that carbetocin is able to activate Gq and in the case of Gi1 and Gi3 promotes an increase in the energy transfer, which could indicate inverse agonism (32), demonstrating a unique functional selective bias towards Gq.
In addition, when compared to oxytocin, carbetocin displays a 5-fold right shifted EC50 value and a 50% reduction in the maximal OXTR/Gq activation value, consistent with a partial agonist profile. These results are in agreement with previous myometrial contractility studies performed in vitro on isolated rat uterine strips reporting an EC50 value of carbetocin (48 nM) about ten times higher than that of oxytocin (5.62 nM) and a maximal contractile responsiveness (Emax) of carbetocin approximately 50% lower than that of oxytocin (34). Substitutions that modify the disulfide bond of oxytocin by introducing a dicarba bond were shown not only to significantly decrease the biological activity of the peptide, but also to induce a loss of receptor selectivity or a switch from an agonist to an antagonist profile (39). Because NMR and crystal structure for carbetocin are currently unavailable, we can only speculate about the molecular basis of these peculiar agonistic properties. In particular, we hypothesize that the modification in the disulfide bond generates distortions in carbetocin secondary structure, inducing different interactions between its cyclic part and the cluster of the residues located in the transmembrane helices TM3, TM5 and TM6 of OXTR. These interactions stabilize different receptor conformations that influence OXTR activation (40) and G-protein coupling selectivity (41) and could be particularly relevant in determining the functional selectivity and partial agonistic properties of oxytocin analogues. Further experiments to investigate carbetocin’s secondary structure, as well as modelling and mutagenesis studies, will be necessary to further address this hypothesis. The unique functional selective OXTR/Gq coupling of carbetocin could be particularly relevant in neuronal cells, where it has been shown that OXTR coupling to Gq and Gi/Go can result in opposite effects on cell excitability via inhibition or activation of potassium channels (42). In this context, carbetocin could be instrumental in identifying the role played by OXTR/Gq coupling in eliciting specific behavioural and neuroendocrine effects, particularly if combined with DNalOVT and atosiban, respectively a Gi1 and Gi3 functional selective agonists previously described (15). Such studies would provide the rationale for pharmacological treatments based on “functional selective agonists” characterised by their specific G-protein isoform coupling. We have previously demonstrated that oxytocin has a strong mitogenic effect when OXTRs are localised in plasma membrane domains enriched in caveolin proteins, glycosphingolipids and cholesterol (lipid rafts), whereas it inhibits cell growth when the receptors are excluded from these domains (43-44). Moreover we found that the inhibition of cell growth is mediate by a pertussis toxin-sensitive G-protein (Gi/o), while the stimulation of cell growth is mediated by a Gi/o-independent pathway, likely via Gq activation (43- 44). Because carbetocin resulted to be an OXTR/Gq functional selective agonist, it may have positive effects on cell proliferation. Experiments using this analogue are currently under way in order to test this hypothesis.
It has been previously demonstrated that carbetocin binds not only to the OXTR but also to vasopressin receptors (34). Competition binding studies performed on rat myometrial and kidney membranes by using tritiated oxytocin and arginine-vasopressin as radiotracers, reported Ki values of 1.96 nM for the rat OXTR, 7.24 nM for the rat V1 receptors (likely V1aR subtype), and 61.3 nM for the rat V2 receptor (34). Despite this previously reported high affinity binding, we did not observe any significant activation of hV1a and hV1b receptors with a concentration of carbetocin as high as 10 μM, suggesting that carbetocin can bind to these receptors without inducing any activation. Moreover, we observed that it can act as a competitive antagonist at the vasopressin receptors. This is an important feature that differentiates carbetocin from oxytocin, as oxytocin is capable to bind to and activate both the V1a and V1b receptors (16, 35). In the past decades a great deal of effort has been devoted to the identification of OXTR selective antagonists and, more recently, the interest in OXTR selective agonists has surged. At present, only one oxytocin peptide analogue, Thr4Gly7OT compound, has proved to be selective for the OXTR, even though its selectivity is limited to mice and rats because Thr4Gly7OT was demonstrated to be rather unselective towards the human oxytocin/V1 receptor subtypes (16, 45). Carbetocin, as a selective OXTR agonist potentially capable to block the vasopressin V1a and V1b receptors represents an interesting agonist worth a careful pharmacological characterisation in vivo, where it may elicit unexpected and at present not fully predicable effects.
Following oxytocin binding, we have previously shown that the OXTR recruited β-arrestinand underwent a rapid (2-10 min) internalisation process followed by receptor recycling to the plasma membrane (36). We report here that, upon carbetocin exposure, instead, the OXTR is efficiently internalised in absence of any significant recruitment of either β-arrestin1 or β-arrestin2. Moreover, once internalised in response to carbetocin, the OXTR remains in the intracellular compartments and does not recycle back to the plasma membrane. Other GPCRs have been shown to be internalised independently of the recruitment of β-arrestins, such as the ghrelin receptor (46) and the serotonin 5HT-2A receptor (47). One possible mechanism for such β-arrestin-independent internalisation relies on the direct binding of an adaptor protein, such as AP2, directly to the OXTR; however, the OXTR lacks the polyarginine motif that is required for direct AP2 interaction (37) making this possibility unlikely. GRK2, a kinase that has been demonstrated to interact with the OXTR (48) can also function as an adaptor protein by interacting directly with clathrin via a clathrin box, mediating a β-arrestins-independent internalisation process (49).
Interestingly, although β-arrestins were not required for carbetocin-induced OXTR internalisation, no OXTR recycling was observed in absence of the recruitment of β-arrestins. It has been previously shown that β-arrestins are fundamental to regulate not only the internalisation but also the recycling of a number of GPCRs. Recycling of GPCRs is thought to occur following endosome acidification, β-arrestin dissociation, and receptor dephosphorylation, a series of processes that occur as the receptor traffics through the vesicular compartments of the cytoplasm. Since arrestins serve as adaptors for many proteins, it is intriguing to speculate that they can specifically regulate the post-endocytotic intracellular trafficking of GPCRs and/or the activity of phosphatases. In particular, it has been demonstrated that in the absence of β-arrestins, the N-formyl peptide receptor undergoes ligand-induced internalisation but is trafficked improperly within the cell, resulting in intracellular retention and impossibility to recycle to the plasma membrane (50). Whatever the mechanism involved, it is relevant that carbetocin allowed to disclose a β-arrestin- independent internalisation pathway of the OXTR, described here for the first time.
The peculiar effects of carbetocin on receptor recycling should be considered when repeated applications are intended to be used for therapeutic purposes, as the absence of receptor recycling could lead to tolerance, a pharmacologically defined phenomenon that consists in a smaller response observed after repeated exposure to a drug (51). Tolerance can be due to the reduction of functional receptor at the plasma membrane, as in the case of an agonist of the motilin receptors, a drug used as gastrointestinal prokinetic agent (52). In the case of octreotide, a somatostatin receptor (SST2R) agonist used for controlling hormone-related symptoms of functioning gastroenteropancreatic neuroendocrine tumours, the antisecretory potency decreased with long-term treatment as a consequence of a persistent internalisation of SST2R (53). Finally, a rebound effect can appear after the discontinuation of a drug (54). There are many classes of medications that produce rebound, including antidepressants, opioids and beta-adrenoreceptor blockers, and in most cases the rebound effect is associated to an up-regulation of receptors (54). Upon these considerations, variations in OXTR expression levels after carbetocin administration should be object of careful future investigation when planning its use in vivo.
CONCLUSIONS
The present study indicates that carbetocin and oxytocin display substantial differences in a number of key molecular pharmacological properties. On one hand, these differences may lead to interesting new effects of carbetocin in comparison to oxytocin. On the other hand, caution should be used to employ carbetocin as a substitute of oxytocin because unexpected (and at present unpredictable) effects could be encountered particularly in vivo. This is especially true for the use of carbetocin in neurodevelopmental and psychiatric conditions, in which the precise role of OXTR and V1 receptors is far from being clear. Further translational studies are needed to understand and fully exploit the therapeutic value of this analogue.
ACKNOWLEDGMENTS
This work was supported by the Telethon Foundation (grant GGP12207), CNR Research Project on Aging and Regione Lombardia (Project MbMM-convenzione n°18099/RCC) to B.C. We thank Dr. C. Galés, INSERM, France who provided the BRET biosensor cDNAs.
COMPETING FINANCIAL INTERESTS
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could represent a potential conflict of interest.
AUTHOR CONTRIBUTION
I.P. performed fluorescence microscopy experiments. M.L. performed fluorescence microscopy experiments. V.G. analysed data and wrote the manuscript. B.C. conceived the project and wrote the manuscript. M.B. conceived and supervised the project, performed BRET and imaging experiments, performed data analysis and wrote the manuscript.
FIGURE LEGENDS
FIGURE 1 Carbetocin selectively activates OXTR/Gq coupling with partial efficacy.
A: Schematic representation of BRET between Rluc8 (the donor) and GFP10 (the acceptor), introduced into the α helical domain of the Gα subunits and the N-terminal domain of Gγ2 (GFP10- Gγ2), respectively. Agonist-induced OXTR-Gα activation leads to a conformational rearrangement of the heterotrimeric G-protein complex that corresponds to a decrease in the BRET ratio. B: BRET was measured in HEK293 cells co-expressing hOXTR, GFP10-Gγ2, Gβ1, and different Rluc8- tagged Gα subunits: Gq, Gi1, Gi2 , Gi3, Go. The results represent the agonist-promoted BRET signal after oxytocin (10 μM) or carbetocin (10 μM) and are expressed as mean values ± SEM. Statistical differences between agonist-promoted BRET in the presence of the indicated Gα proteins and PBS-treated controls (dotted line) were determined by one-way ANOVA followed by Dunnett’s post-hoc test (*p <0.05, **p <0.01, ***p <0.001). C: Cells were transfected with hOXTR, GFP10- Gγ2, Gβ1, Gαq-Rluc8 and were stimulated with different concentrations of oxytocin or carbetocin (from 10-13 to 10-5 M) for 2 min. The results represent the “BRET ligand effect” signals for oxytocin and carbetocin and are expressed as the mean value ± SEM of at least three independent experiments, each performed at least in duplicate.
FIGURE 2 Carbetocin does not activate and acts as competitive antagonist for vasopressin V1a and V1b receptors
A: V1aR or B: V1bR were transfected together with GFP10-Gγ2, Gβ1, and different Rluc8-tagged Gα subunits: Gq, Gi1, Gi2, Gi3, Go. The results represent the agonist-induced BRET signal with oxytocin (10 μM), carbetocin (10 μM) or arginine-vasopressin (10 μM) and are expressed as mean values ± SEM of at least three independent experiments, each performed at least in duplicate. Statistical differences between “BRET ligand effect” signal in the presence of the indicated Gα proteins and PBS-treated controls (dotted line) were determined by one-way ANOVA followed by Dunnett’s post-hoc test (*p<0.05, ** p<0.01,***p<0.001). Cells transfected with Gαq-Rluc, GFP10-Gγ2, Gβ1 and C: V1aR or D: V1bR were stimulated with increasing concentrations of arginine-vasopressin alone (blue line) or simultaneously treated with carbetocin 1μM (red dotted line) or 10 μM (red solid line) for 2 min. The results represent the “BRET ligand effect” signals for arginine-vasopressin and arginine-vasopressin + carbetocin and are expressed as the mean value ± SEM of at least three independent experiments, each performed at least in duplicate.
FIGURE 3 Carbetocin does not promote the recruitment of β-arrestins.
A: Schematic representation of BRET between Rluc and Venus introduced at the C-terminus of hOXTR (hOXTR-RLuc) and of the βarrestins (βarrestin-Venus), respectively. HEK293 cells co- expressing hOXTR-Rluc and B: βarrestin1-Venus or C: βarrestin2-Venus were stimulated by maximal doses (10 μM) of oxytocin and carbetocin. BRET measurements were made in real-time for 5 min. The results represent the “BRET ligand effect” signal for oxytocin and carbetocin and are expressed as the mean value ± SEM of a representative experiment (from three independent experiments) performed in duplicate. The curve was generated as one-phase association.
FIGURE 4 Carbetocin promotes OXTR internalisation independently from the recruitment of β-arrestins.
Representative confocal microscopy images of cells transfected with hOXTR-RFP (red) and A: β- arrestin1-Venus (green) and B: β-arrestin2-Venus (green) treated with PBS (Basal), oxytocin (100 nM) or carbetocin (1 μM) for 2 min. After agonist stimulation cells were fixed, incubated with DAPI to stain nuclei and imaged by confocal microscopy. Pictures shown are representative of 3 independent experiments. Scale bar = 10μm
FIGURE 5 Carbetocin treatment prevents OXTR recycling to the plasma membrane after agonist removal.
Representative confocal microscopy images of cells transfected with hOXTR-RFP (red) A: at basal conditions and B: after 15 min oxytocin (100 nM) or carbetocin (1 μM). C: In recycling experiments, cells were treated for 15 min with oxytocin (100 nM) or carbetocin (1 μM), washed in acidic buffer, and kept in the absence of agonists for 4 h. They were then fixed, incubated with DAPI to stain nuclei and imaged by confocal microscopy. Pictures shown are representative of 3 independent experiments. Scale bar = 10μm
REFERENCES
-
Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, Osten P, Schwarz MK, Seeburg PH, Stoop R et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 2012, 73:553-566.
-
Leng G, Ludwig M. Neurotransmitters and peptides: whispered secrets and public announcements. J Physiol 2008, 586:5625-5632.
-
Arrowsmith S, Wray S. Oxytocin: its mechanism of action and receptor signalling in the myometrium. J Neuroendocrinol 2014, 26:356-369.
-
Dahlke JD, Mendez-Figueroa H, Maggio L, Hauspurg AK, Sperling JD, Chauhan SP, Rouse DJ. Prevention and management of postpartum hemorrhage: a comparison of 4 national guidelines.Am J Obstet Gynecol 2015, 213:76 e71-10.
Crowley WR. Neuroendocrine regulation of lactation and milk production. Compr Physiol 2015,
5:255-291.
-
Young LJ. Oxytocin, social cognition and psychiatry. Neuropsychopharmacology 2015, 40:243-
244.
-
Churchland PS, Winkielman P. Modulating social behavior with oxytocin: how does it work?
What does it mean? Horm Behav 2012, 61:392-399.
-
Guastella AJ, Hickie IB. Oxytocin Treatment, Circuitry and Autism: A Critical Review of the
Literature Placing Oxytocin into the Autism Context. Biol Psychiatry 2015,
-
Feifel D, Shilling PD, MacDonald K. A Review of Oxytocin's Effects on the Positive, Negative, and
Cognitive Domains of Schizophrenia. Biol Psychiatry 2015,
-
Bakermans-Kranenburg MJ, van IJMH. Sniffing around oxytocin: review and meta-analyses of
trials in healthy and clinical groups with implications for pharmacotherapy. Transl Psychiatry
2013, 3:e258.
-
Gazis D. Plasma half-lives of vasopressin and oxytocin analogs after iv injection in rats. Proc Soc
Exp Biol Med 1978, 158:663-665.
-
Mens WB, Laczi F, Tonnaer JA, de Kloet ER, van Wimersma Greidanus TB. Vasopressin and
oxytocin content in cerebrospinal fluid and in various brain areas after administration of
histamine and pentylenetetrazol. Pharmacol Biochem Behav 1983, 19:587-591.
-
Leng G, Ludwig M. Intranasal Oxytocin: Myths and Delusions. Biol Psychiatry 2015,
-
Pittman QJ, Spencer SJ. Neurohypophysial peptides: gatekeepers in the amygdala. Trends
Endocrinol Metab 2005, 16:343-344.
-
Busnelli M, Sauliere A, Manning M, Bouvier M, Gales C, Chini B. Functional selective oxytocin-
derived agonists discriminate between individual G protein family subtypes. J Biol Chem 2012,
287:3617-3629.
-
Busnelli M, Bulgheroni E, Manning M, Kleinau G, Chini B. Selective and potent agonists and
antagonists for investigating the role of mouse oxytocin receptors. J Pharmacol Exp Ther 2013,
346:318-327.
-
Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, Durroux T, Mouillac B, Corbani M,
Guillon G. Oxytocin and vasopressin agonists and antagonists as research tools and potential
therapeutics. J Neuroendocrinol 2012, 24:609-628.
-
Barth T, Krejci I, Vaneckova J, Jost K, Rychlik I. Prolonged action of deamino-carba analogues of
oxytocin on the rat uterus in vivo. Eur J Pharmacol 1974, 25:67-70.
-
Cort N, Einarsson S, Schams D, Vilhardt H. Blood concentrations of oxytocin equivalents after
single injections of deamino-1-monocarba-[2-O-methyltyrosine]-oxytocin in lactating sows. Am J
Vet Res 1981, 42:1804-1806.
-
Cort N, Einarsson S, Viring S. Actions of oxytocin and a long-acting carba oxytocin analog on the
porcine myometrium in vitro and in vivo. Am J Vet Res 1979, 40:430-432.
-
Cort N, Einarsson S, Astrom G. Effect of oxytocin and its long-acting analog on milk let-down and
intramammary pressure in healthy lactating sows. Am J Vet Res 1982, 43:1283-1285.
-
Amsalem H, Aldrich CJ, Oskamp M, Windrim R, Farine D. Postpartum uterine response to
oxytocin and carbetocin. J Reprod Med 2014, 59:167-173.
-
Mak P, Broussard C, Vacy K, Broadbear JH. Modulation of anxiety behavior in the elevated plus
maze using peptidic oxytocin and vasopressin receptor ligands in the rat. J Psychopharmacol
2012, 26:532-542.
-
Chaviaras S, Mak P, Ralph D, Krishnan L, Broadbear JH. Assessing the antidepressant-like effects
of carbetocin, an oxytocin agonist, using a modification of the forced swimming test.
Psychopharmacology (Berl) 2010, 210:35-43.
-
Zanos P, Georgiou P, Wright SR, Hourani SM, Kitchen I, Winsky-Sommerer R, Bailey A. The
oxytocin analogue carbetocin prevents emotional impairment and stress-induced reinstatement
of opioid-seeking in morphine-abstinent mice. Neuropsychopharmacology 2014, 39:855-865.
-
Georgiou P, Zanos P, Ehteramyan M, Hourani S, Kitchen I, Maldonado R, Bailey A. Differential regulation of mGlu5 R and MuOPr by priming- and cue-induced reinstatement of cocaine-seeking behaviour in mice. Addict Biol 2015, 20:902-912.
Feifel D, Shilling PD, Belcher AM. The effects of oxytocin and its analog, carbetocin, on genetic
deficits in sensorimotor gating. Eur Neuropsychopharmacol 2012, 22:374-378.
-
Klenerova V, Krejci I, Sida P, Hlinak Z, Hynie S. Oxytocin and carbetocin effects on spontaneous behavior of male rats: modulation by oxytocin receptor antagonists. Neuro Endocrinol Lett 2009,
30:335-342.
-
Klenerova V, Krejci I, Sida P, Hlinak Z, Hynie S. Modulary effects of oxytocin and carbetocin on
stress-induced changes in rat behavior in the open-field. J Physiol Pharmacol 2009, 60:57-62.
-
Klenerova V, Krejci I, Sida P, Hlinak Z, Hynie S. Oxytocin and carbetocin ameliorating effects on restraint stress-induced short- and long-term behavioral changes in rats. Neuro Endocrinol Lett
2010, 31:622-630.
-
Sauliere A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altie MF, Seguelas MH, Pathak A,
Hansen JL et al. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity.
Nat Chem Biol 2012, 8:622-630.
-
M'Kadmi C, Leyris JP, Onfroy L, Gales C, Sauliere A, Gagne D, Damian M, Mary S, Maingot M,
Denoyelle S et al. Agonism, Antagonism, and Inverse Agonism Bias at the Ghrelin Receptor
Signaling. J Biol Chem 2015, 290:27021-27039.
-
Atke A, Vilhardt H. Uterotonic activity and myometrial receptor affinity of 1-deamino-1-carba-2-
tyrosine(O-methyl)-oxytocin. Acta Endocrinol (Copenh) 1987, 115:155-160.
-
Engstrom T, Barth T, Melin P, Vilhardt H. Oxytocin receptor binding and uterotonic activity of carbetocin and its metabolites following enzymatic degradation. Eur J Pharmacol 1998, 355:203-
210.
-
Wisniewski K, Alagarsamy S, Galyean R, Tariga H, Thompson D, Ly B, Wisniewska H, Qi S, Croston
G, Laporte R et al. New, potent, and selective peptidic oxytocin receptor agonists. J Med Chem
2014, 57:5306-5317.
-
Conti F, Sertic S, Reversi A, Chini B. Intracellular trafficking of the human oxytocin receptor:
evidence of receptor recycling via a Rab4/Rab5 "short cycle". Am J Physiol Endocrinol Metab
2009, 296:E532-542.
-
Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis.
Traffic 2007, 8:462-470.
-
Gimpl G, Postina R, Fahrenholz F, Reinheimer T. Binding domains of the oxytocin receptor for the
selective oxytocin receptor antagonist barusiban in comparison to the agonists oxytocin and
carbetocin. Eur J Pharmacol 2005, 510:9-16.
-
Stymiest JL, Mitchell BF, Wong S, Vederas JC. Synthesis of biologically active dicarba analogues of
the peptide hormone oxytocin using ring-closing metathesis. Org Lett 2003, 5:47-49.
-
Chini B, Mouillac B, Balestre MN, Trumpp-Kallmeyer S, Hoflack J, Hibert M, Andriolo M, Pupier S, Jard S, Barberis C. Two aromatic residues regulate the response of the human oxytocin receptor
to the partial agonist arginine vasopressin. FEBS Lett 1996, 397:201-206.
-
Favre N, Fanelli F, Missotten M, Nichols A, Wilson J, di Tiani M, Rommel C, Scheer A. The DRY
motif as a molecular switch of the human oxytocin receptor. Biochemistry 2005, 44:9990-10008.
-
Gravati M, Busnelli M, Bulgheroni E, Reversi A, Spaiardi P, Parenti M, Toselli M, Chini B. Dual modulation of inward rectifier potassium currents in olfactory neuronal cells by promiscuous G
protein coupling of the oxytocin receptor. J Neurochem 2010, 114:1424-1435.
-
Guzzi F, Zanchetta D, Cassoni P, Guzzi V, Francolini M, Parenti M, Chini B. Localization of the human oxytocin receptor in caveolin-1 enriched domains turns the receptor-mediated inhibition
of cell growth into a proliferative response. Oncogene 2002, 21:1658-1667.
-
Reversi A, Rimoldi V, Brambillasca S, Chini B. Effects of cholesterol manipulation on the signaling
of the human oxytocin receptor. Am J Physiol Regul Integr Comp Physiol 2006, 291:R861-869.
-
Guillon G, Derick S, Pena A, Cheng LL, Stoev S, Seyer R, Morgat JL, Barberis C, Gal CS, Wagnon J et al. The discovery of novel vasopressin V1b receptor ligands for pharmacological, functional and
structural investigations. J Neuroendocrinol 2004, 16:356-361.
-
Holliday ND, Holst B, Rodionova EA, Schwartz TW, Cox HM. Importance of constitutive activity and arrestin-independent mechanisms for intracellular trafficking of the ghrelin receptor. MolEndocrinol 2007, 21:3100-3112.
Gray JA, Bhatnagar A, Gurevich VV, Roth BL. The interaction of a constitutively active arrestin
with the arrestin-insensitive 5-HT(2A) receptor induces agonist-independent internalization. Mol
Pharmacol 2003, 63:961-972.
-
Hasbi A, Devost D, Laporte SA, Zingg HH. Real-time detection of interactions between the human
oxytocin receptor and G protein-coupled receptor kinase-2. Mol Endocrinol 2004, 18:1277-1286.
-
Shiina T, Arai K, Tanabe S, Yoshida N, Haga T, Nagao T, Kurose H. Clathrin box in G protein-
coupled receptor kinase 2. J Biol Chem 2001, 276:33019-33026.
-
Vines CM, Revankar CM, Maestas DC, LaRusch LL, Cimino DF, Kohout TA, Lefkowitz RJ, Prossnitz
ER. N-formyl peptide receptors internalize but do not recycle in the absence of arrestins. J Biol
Chem 2003, 278:41581-41584.
-
Colpaert FC. Drug discrimination: no evidence for tolerance to opiates. Pharmacol Rev 1995,
47:605-629.
-
Lamian V, Rich A, Ma Z, Li J, Seethala R, Gordon D, Dubaquie Y. Characterization of agonist-
induced motilin receptor trafficking and its implications for tachyphylaxis. Mol Pharmacol 2006,
69:109-118.
-
Mohamed A, Blanchard MP, Albertelli M, Barbieri F, Brue T, Niccoli P, Delpero JR, Monges G,
Garcia S, Ferone D et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in
human pancreatic neuroendocrine tumor cultures. Endocr Relat Cancer 2014, 21:691-704.
-
Reidenberg MM. Drug discontinuation effects are part of the pharmacology of a drug. J
Pharmacol Exp Ther 2011, 339:324-328.